
4 Système de management de la qualité F 22 .gif)
4.1 Exigences générales
Exigences générales, développer un SMQ, processus, modifications, maîtrise des processus externalisés, valider les logiciels
Exigences 1 à 26 (voir aussi le quiz)

Dans le schéma simplifié de la figure 4-1 on peut voir la finalité d'un système de management de la qualité ISO 13485 :

Figure 4-1. Finalité d'un SMQ ISO 13485
Les exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) de la norme ISO 13485 sont montrées en figure 4-2 :
.jpg)
Figure 4-2. Les exigences de la norme ISO 13485
Les exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) liées aux produits sont spécifiées par le clientcelui qui reçoit un produit (voir aussi ISO 9000, 3.3.5), par l'entreprisestructure qui satisfait un besoin (voir aussi ISO/IEC Guide 2, 4.2 et ISO 26 000, 2.12) ou une réglementation.
Les exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) de la norme ISO 13485 concernent exclusivement le système de management de la qualité et ses processusactivités qui transforment des éléments d'entrée en éléments de sortie (voir aussi ISO 9000, 3.4.1) :
- les exigences des clients et les exigences réglementaires applicables sont identifiées et prises en compte
- le système de management de la qualité (SMQ) est établi, documenté, mis en œuvre, et maintenu
- la politique qualité, les objectifs, les ressources et l'environnement de travail sont déterminés
- les rôles du fabricant, de l’importateur, du distributeur et du représentant autorisé sont documentés

- les processus nécessaires au SMQ :
- sont identifiés, mesurés, surveillés
- les enregistrements sont conservés relatifs au respect des exigences :
- de la norme ISO 13485
- réglementaires applicables
- leurs objectifs établis et analysés
- leur séquence et les interactions déterminées
- leurs ressources nécessaires fournies
- leurs critères opérationnels établis
- leur information indispensable pour la surveillance assurée
- leurs pilotes nommés
- leurs enregistrements requis conservés
- le niveau de risque des processus est identifié par rapport à :
- l'impact sur la sécurité et les performances des dispositifs médicaux
- la conformité réglementaire
- les actions pour atteindre les résultats planifiés et maintenir l'efficacité des processus sont établies et mises en œuvre
- le personnel est impliqué
- évaluées par rapport à leur impact sur le SMQ
- évaluées par rapport à leur impact sur les dispositifs médicaux
- maîtrisés conformément aux exigences :
- de la norme ISO 13485
- réglementaires applicables
La procédure de validation des logiciels inclut : 
- la validation des applications logicielles avant première utilisation
- la revalidation après modification du logiciel ou de l’application
- l'approche proportionnée au risque associé à l’utilisation
- la conservation des enregistrements
.jpg)
- faire de la sur-qualité :
- une opération inutile est réalisée sans que cela ajoute de la valeur et sans que le client le demande – c’est un gaspillage, cf. les outils qualité E 12
- faire écrire toutes les procédures par le responsable qualité :
.jpg)
- la qualité est l'affaire de tous, cf. le paragraphe 6.2 – « le personnel a conscience de la pertinence et de l’importance de chacun à la contribution aux objectifs qualité », ce qui est encore plus vrai pour les chefs de départements et les pilotes de processus
- oublier les spécificités liées à la culture de l'entreprise :
- innovation, luxe, secret, management autoritaire (Apple)
- culture forte liée à l’écologie, à l’action et la lutte, tout en cultivant le secret (Greenpeace)
- culture d’entreprise fun et décalée (Michel&Augustin)
- entreprise libérée, l’homme est bon, aimer son client, rêve partagé (Favi, cf. F 50)
- la cartographie des processus contient assez de flèches pour bien montrer qui est le client (interne ou externe)
- la valeur ajoutée du processus est dévoilée pendant la revue de processus
- la liste des processus est à jour
- l’analyse de la performance des processus est un exemple de preuve du maintien de l’efficacité du SMQ
- le rôle de l’entreprise est documenté selon les exigences réglementaires applicables
- les incidences des modifications des processus sont évaluées
- des contrats qualité sont établis pour les processus externalisés critiques
- les enregistrements de la validation des applications logicielles sont codifiés et conservés
- la finalité de chaque processus est clairement définie
- certains éléments de sortie de processus ne sont pas correctement définis (clients non pris en compte)
- critères d’efficacité des processus non établis
- liste des processus non actualisée
- le rôle de l’entreprise n’est pas documenté
- pilote de processus non formalisé
- processus externalisés non identifiés
- des activités bien réelles ne sont identifiées dans aucun processus
- maîtrise des prestations externalisées non décrite
- séquences et interactions de certains processus ne sont pas identifiées
- critères et méthodes pour assurer l’efficacité des processus non définis
- surveillance de l’efficacité de certains processus non établie
- l’incidence des modifications des processus n’est pas évaluée
- des applications logicielles ne sont pas validées
4.2 Documentation
Exigences relatives à la documentation, manuel qualité, dossier du dispositif médical, maîtrise des documents et des enregistrements
Exigences 27 à 62

Le bon document, au bon endroit, au bon moment
La documentation du SMQsystème de management de la qualité (cf. figure 4-3) comprend :
- le manuel qualité (MQ)
- la politique qualité
- les objectifs qualité
- les fiches processus
- les procédures
- les documents nécessaires pour maîtriser les processus
- les documents d’origine externe (des fournisseurs, des clients, les normes)
- les documents requis par les exigences réglementaires applicables
- les enregistrements exigés
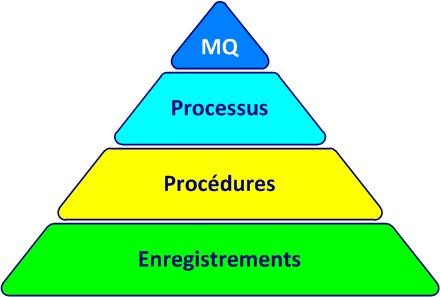
Figure 4-3. La pyramide documentaire
Dans le manuel qualité (cf. annexe 08) sont décrits :
- le domaine d’application du SMQ
- les types de dispositifs médicaux fabriqués
- les procédures (cf. annexe 09) ou une référence à celles-ci
- la séquence et les interactions entre les processus (cartographie des processus, cf. § 3.3, annexe 03)
- la justification des exclusions d'éléments des articles 6, 7 et 8 (cf. § 1.2)
Le manuel qualité c’est comme le code de la route, c’est surtout un guide, un outil, mais il ne vous apprend pas à conduire.
Le dossier du dispositif médical comprend la documentation technique développée et maintenue pour chaque modèle ou type de DM et chaque prestation associée. Cette documentation comprend :
- la description du produit, usage prévu, étiquette, instructions d'utilisation
- la nomenclature :
- ensembles, sous-ensembles
- composants
- matériaux
- étiquettes, instructions d’utilisation, fiche d’avertissement
- accessoires
- emballages
- la classification et exigences réglementaires applicables
- le processus de fabrication, conditionnement, stockage, manutention et distribution (y compris les méthodes de stérilisation, inspections, essais et validations)
- les diagrammes de flux, les plans, dessins, diagrammes d’assemblage, les instructions de travail
- les procédures de mesure et de surveillance
- l’analyse des risques
- les exigences d’installation, si approprié
- les procédures de prestations associées, si approprié
- les résultats des vérifications, validations, essais, tests, les données cliniques, la déclaration de conformité
Les documents nécessitant une approbation des modifications par les clientscelui qui reçoit un produit (voir aussi ISO 9000, 3.3.5) ou autorités sont identifiés.
Un tableau regroupant les exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) des clientscelui qui reçoit un produit (voir aussi ISO 9000, 3.3.5), les exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) réglementaires applicables et les processusactivités qui transforment des éléments d'entrée en éléments de sortie (voir aussi ISO 9000, 3.4.1) est d’une grande aide pour vérifier la conformité du SMQsystème de management de la qualité.
Chaque document interne est vérifié et approuvé. Toute procédure, exigencebesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) ou activité documentée est mise en œuvre et tenue à jour. Les documents périmés (obsolètes) sont identifiés, conservés et leur utilisation interdite en atelier.
L'avant dernière version d'un document peut devenir un enregistrement.
La norme ISO 10013 (2021) : "Systèmes de management de la qualité - Recommandations pour les informations documentées" fournit des recommandations par rapport à la documentation d'un SMQsystème de management de la qualité.
Des réponses aux 416 exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) (dans le texte "doit/doivent" en anglais "shall") des articles 4 à 8 de la norme ISO 13485 sont présentes dans la documentation. Les exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) de l’ISO 9001 « ne sont » que 305.
Procédure : ensemble d’actions à entreprendre pour effectuer un processus
Manuel qualité : document énonçant les dispositions générales d’une entreprise pour obtenir des produits ou services conformes
Document : tout support permettant le traitement d’une information
Enregistrement : document fournissant des preuves tangibles des résultats obtenus
Les procédures de l'ISO 13485 (cf. annexe 09) sont :
- Validation des logiciels (§ 4.1.6)
-
Maîtrise des documents (§ 4.2.4). La procédure assure :
- la vérification (revue du contenu et de la forme)
- l’approbation (ensuite autorisation de diffusion)
- la mise à jour (vérification et approbation de nouveau)
- l'identification de la version pertinente en vigueur à l'endroit d'utilisation
- la lisibilité
- a disponibilité
- l'identification et la distribution des documents d'origine externe
- l'empêchement d’utilisation de documents périmés (obsolètes) et leur identification spécifique
- la prévention de la perte ou détérioration des documents
- la conservation de documents périmés des dispositifs médicaux
- Maîtrise des enregistrements (§ 4.2.5). La procédure assure :
- l’identification
- le stockage
- la sécurité
- l’intégrité
- la récupération
- la disponibilité
- la durée de conservation
- l’élimination
- la lisibilité
- la diffusion
- l’identification des modifications
- Revue de direction (§ 5.6)
- Maîtrise de l'environnement de travail (§ 6.4.1)
- Conception et développement (§ 7.3)
- Transfert de la conception et du développement (§ 7.3.8)
- Maîtrise des modifications (§ 7.3.9)
- Achats (§ 7.4)
- Maîtrise de la production (§ 7.5.1 et 8.2.6)
- Prestations associées (§ 7.5.4)
- Validation des processus (§ 7.5.6)
- Identification et traçabilité (§§ 7.5.8 et 7.5.9)
- Préservation du produit (§ 7.5.11)
- Equipements de surveillance et de mesure (§ 7.6)
- Retours d'information (§ 8.2.1)
- Traitement des réclamations (§ 8.2.2)
- Surveillance après commercialisation (§ 8.2)
- Veille réglementaire (§ 8.2)
- Signalement aux autorités réglementaires (§ 8.2.3)
- Audit interne (§ 8.2.4)
- Maîtrise du produit non conforme (§ 8.3.1)
- Fiches d'avertissement (§ 8.3.3)
- Retouches (§ 8.3.4)
- Analyse des données (§ 8.4)
- Action corrective (§ 8.5.2)
- Action préventive (§ 8.5.3)
On peut regrouper plusieurs procédures en une seule. La documentation peut être sous toute forme et tout type de support. Elle contribue entre autres à fournir des preuves tangibles et évaluer l’efficacité et la pertinence du SMQsystème de management de la qualité.
Preuve tangible : donnée factuelle dont la véracité peut être démontrée
Retouche (reprise) : action sur un produit pour le rendre conforme
Une procédure peut être documentée ou ne pas l'être : (cf. ISO 9000 : 2015, § 3.4.5, note 1 : "Les procédures peuvent ou non faire l’objet d’un document"). Notre préférence est pour la solution documentée (écrite), courte, simple et pertinente. Surtout dans les cas ou l’absence de procédure peut entraîner des écarts de la politique, des objectifs qualité, de la sécurité ou des performances du dispositif médical.
Les modifications effectuées sont réalisées par l’auteur du document et revues et approuvées par une personne ayant les informations pertinentes pour prendre ses décisions.
Des méthodes de protection des enregistrements contenant des informations confidentielles de santé sont mises en place conformément aux exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) réglementaires applicables.
Une revue de la documentation est conduite périodiquement par le responsable qualité.
La période de conservation d’au moins une copie des documents est déterminée (procédures et enregistrements). Cette période respecte les exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) réglementaires applicables et ne peut être inférieure à la fin de durée de vie du dispositif médical et d’au moins deux ans de la sortie sur le marché du DM.
La documentation est liée à la taille et type de l'entreprisestructure qui satisfait un besoin (voir aussi ISO/IEC Guide 2, 4.2 et ISO 26 000, 2.12), à la complexité des processusactivités qui transforment des éléments d'entrée en éléments de sortie (voir aussi ISO 9000, 3.4.1) et à la compétence du personnel. Cette documentation est accessible au personnel concerné et il en est informé lors d’évolutions. Seuls les documents strictement nécessaires sont obligatoires pour obtenir une documentation simplifiée.
Exemple de documents couramment utilisés :
- manuel qualité
- organigramme de l’entreprise
- procédures
- plans qualité
- spécifications
- instructions de travail ou d'essais
- formulaires
- enregistrements
- AMDEC
- documents externes
- liste de fournisseurs approuvés
- plans d'essais et d'inspection
Spécification : description définitive des exigences d’un système ou produit dans le but de le développer ou de le valider
Le processusactivités qui transforment des éléments d'entrée en éléments de sortie (voir aussi ISO 9000, 3.4.1) « gérer la documentation » assure la revue, la diffusion, les modifications et la mise en œuvre des spécifications techniques du clientcelui qui reçoit un produit (voir aussi ISO 9000, 3.3.5). .jpg)
Les paroles s'envolent, les écrits restent. Proverbe latin
Enregistrements conservés requis par l'ISO 13 485 et le règlement 2017/745pour prouver la conformité et l’efficacité du SMQsystème de management de la qualité (paragraphes) :
- rôle de l'entreprise (4.1.1)
- rapport bénéfice/risque (4.1)
- maîtrise des processus (4.1.3 e et 4.2.1 d)
- validation des applications logicielles (4.1.6)
- documentation générale du SMQ (4.2.1)
- exigences réglementaires (4.2.1 e)
- dossier du dispositif médical (4.2.3)
- maîtrise des enregistrements (4.2.4)
- responsabilités, autorités et indépendance (5.5.1)
- revue de direction (5.6.1 et 5.6.3)
- compétences du personnel (6.2 e)
- maintenance des infrastructures (6.3)
- environnement de travail (6.4.1)
- maîtrise de la contamination (6.4.2)
- gestion des risques (7.1)
- planification de la production (7.1)
- conformité du processus et du produit (7.1 d)
- revue des exigences relatives au produit (7.2.2)
- communication avec le client (7.2.3)
- éléments d'entrée de la conception et du développement (7.3.3)
- éléments de sortie de la conception et du développement (7.3.4)
- revue de la conception et du développement (7.3.5)
- vérification de la conception et du développement (7.3.6)
- validation de la conception et du développement (7.3.7)
- transfert vers la fabrication de la conception et du développement (7.3.8)
- modifications de la conception et du développement (7.3.9)
- dossiers de la conception et du développement (7.3.10)
- maîtrise des fournisseurs (7.4.1)
- informations avec les fournisseurs (7.4.2)
- vérification du produit acheté (7.4.3)
- vérification et approbation des dispositifs médicaux avant libération (7.5.1)
- propreté du produit (7.5.2)
- installation et vérification des dispositifs médicaux (7.5.3)
- prestations associées réalisées (7.5.4)
- paramètres de stérilisation des lots (7.5.5)
- validation des processus (7.5.6)
- validation des procédés de stérilisation (7.5.7)
- identification unique (7.5.8)
- traçabilité (7.5.9.1)
- destinataire de l'emballage (7.5.9.2)
- souci avec la propriété du client (7.5.10)
- préservation du produit (7.5.11)
- étalonnage et vérification des équipements de mesure (7.6)
- résultats de validation des logiciels de surveillance et de mesure (7.6)
- retour d'information (8.2.1)
- traitement des réclamations (8.2.2)
- revue de la législation (8.2)
- inventaire des exigences légales (8.2)
- plan de surveillance après commercialisation (8.2)
- rapport de surveillance après commercialisaton (8.2)
- signalement aux autorités réglementaires (8.2.3)
- audits internes (8.2.4)
- surveillance et mesure du produit (8.2.6)
- non-conformités (8.3.1)
- autorisation de dérogation (8.3.2)
- fiches d'avertissement (8.3.3)
- retouches réalisées (8.3.4)
- analyse des données (8.4)
- actions correctives entreprises (8.5.2)
- actions préventives entreprises (8.5.3)
Chaque enregistrement est unique et normalement ne peut être modifié, sauf pour correction d'erreur. Tout enregistrement apporte la preuve d'une tâche, d'une opération, d'une activité, d'un processusactivités qui transforment des éléments d'entrée en éléments de sortie (voir aussi ISO 9000, 3.4.1) ou d'une exigencebesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1). Les enregistrements sont la base de données indispensable pour analyser l'efficacité des processusactivités qui transforment des éléments d'entrée en éléments de sortie (voir aussi ISO 9000, 3.4.1) et contribuer au maintien du SMQ. Exemples d'autres enregistrements souvent utilisés :
- fiche d'avertissement
- étude de capabilité processus
- coûts d'obtention de la qualité
- demande de modification
- demande de dérogation
- plainte du client
- bon de livraison
- fiche de non-conformité
- attestation de conformité
La période de conservation des enregistrements est déterminée et d’habitude est incluse dans la liste principale des documents. Cette période est au moins de deux ans, respecte les exigencesbesoin ou attente implicites ou explicites (voir aussi ISO 9000, 3.1.2 et 3.12.1) réglementaires applicables et ne peut être inférieure à la durée de vie du dispositif médical.
- le manuel qualité est court et simplifié (facile à lire par l’ensemble du personnel). Il contient le domaine d'application et la justification des exclusions
- la gestion des documents permet de montrer clairement l’auteur, le vérificateur et l’approbateur du document initial et des versions suivantes
- la gestion des modifications d’un document (ligne au milieu de l’ancien texte, couleur rouge) permet de voir rapidement l’historique
- la liste des dates de mise en œuvre des modifications en production est accessible en atelier
- le dossier de chaque type de dispositif médical est complet
- les modes de diffusion des documents sont décrits dans la procédure « Maîtrise des documents »
- la hiérarchisation des documents est logique et claire (manuel, procédures, processus, instructions, enregistrements)
- la revue de toute la documentation du SMQ, conduite deux fois par an, est très bien organisée, les actions sont finalisées dans les délais fixés
- la liste générale des documents contient aussi la durée de conservation des enregistrements
- la durée de conservation des enregistrements respecte les exigences réglementaires applicables
- les documents externes (normes, règlements applicables, documents clients, fournisseurs et machines) sont codifiés comme les documents internes et dans une liste spécifique l’endroit de rangement de chaque document externe est notifié
- le manuel qualité n’est pas à jour
- dans le manuel qualité les exclusions ne sont ni détaillées ni justifiées
- des procédures ne sont pas à jour
- la nécessité de certains documents n’est pas évaluée
- des activités bien réelles ne sont identifiées dans aucun document
- certains documents ne sont pas codifiés
- les dossiers de certains dispositifs médicaux ne sont pas complets
- des documents ne sont pas à l’endroit où l’on en a besoin
- des instructions ne sont pas à jour (avant dernière version)
- pas de mesures pour assurer la sécurité des enregistrements
- des modifications aux enregistrements sont difficilement identifiables
- des modifications ne sont pas approuvées par les personnes ayant l’autorité
- des documents ne sont pas approuvés avant leur diffusion
- lors de la réunion de lancement d’un projet la liste des participants n’est pas enregistrée
- la protection des documents sur le réseau n’est pas définie
- les documents externes (client, fournisseur) ne sont pas maîtrisés (codifiés)
- la durée de conservation et les modalités d’élimination des enregistrements ne sont pas établies
.jpg) Minute de détente. Jeu : Documentation
Minute de détente. Jeu : Documentation
Le reste de la formation F 22v16 Préparation à l'ISO 13485 version 2016 est accessible sur cette page.
Voir aussi la formation F 42v16 Audit interne ISO 13485 et le lot de formations ISO 13485.